ChimeraX Recipes
Open multi-file AlphaFold models
AlphaFold models larger than 1400 amino acids are computed in multiple segments each 1400 amino acids long spaced every 200 amino acids. This is the longest sequence length the method can handle due to GPU memory limits. Here is Python code to load the segments from separate files and align them. Opening the Python defines the bigalpha command
open bigalpha.py
You will need to download the human AlphaFold database predicted structures (5 Gbytes) from EBI. Then open the segments for protein titin (UniProt Q8WZ42, 34350 amino acids) using ChimeraX command
bigalpha Q8WZ42 directory /directory/of/alphafold/models
Segments every 1200 amino acids are loaded each segment aligned to the last 5 residues (C-alpha atoms) of the preceding segment and the 200 overlap residues in the added segment are deleted.
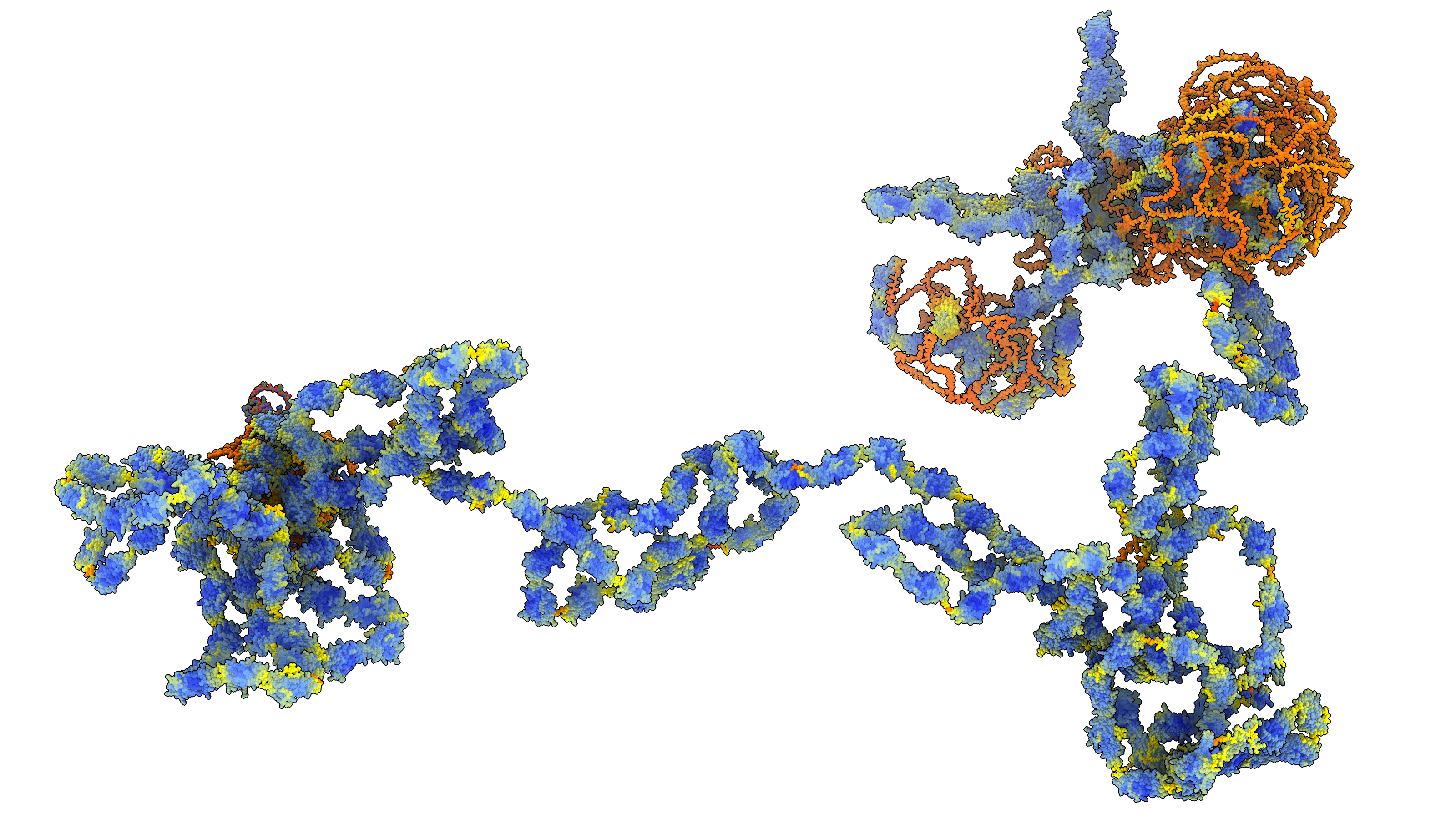
Here is the bigalpha.py code:
#
# Open AlphaFold database models for proteins larger than 1400 amino acids.
# These calculated in 1400 amino acid segments every 200 amino acids due to
# limitations (GPU memory) of the AlphaFold software. We load and align
# the segment models. This produces many clashes.
#
# Opening this Python in ChimeraX (version newer than August 2021 because it
# uses the new combine command) will register the "bigalpha" command.
# You need to have downloaded the human AlphaFold database models from
#
# https://alphafold.ebi.ac.uk/download
#
# Then ChimeraX commands to load the model of the protein Titin
#
# cd /directory/of/alphafold/models
# bigalpha Q8WZ42
#
def open_multifile_alphafold_model(session, uniprot_id = 'Q8WZ42', directory = '.',
combine = True, residues_per_file = 1400,
overlap = 200, align_span = 5):
# Allow multiple UniProt identifiers comma-separated.
if ',' in uniprot_id:
# Handle multiple uniprot ids
models = []
for uid in uniprot_id.split(','):
m = open_multifile_alphafold_model(session, uid, directory, combine,
residues_per_file, overlap)
models.extend(m)
return models
# Find the AlphaFold structure files for this UniProt identifier.
from os import listdir
all_filenames = listdir(directory)
filenames = [filename for filename in all_filenames
if filename.startswith('AF-%s' % uniprot_id)
and filename.endswith('.pdb.gz')]
nfiles = len(filenames)
print ('AlphaFold %s is split into %d mmCIF files' % (uniprot_id, nfiles))
# Cannot read compressed mmCIF directly
# filename = 'AF-%s-F%%d-model_v1.cif.gz' % uniprot_id
filename = 'AF-%s-F%%d-model_v1.pdb.gz' % uniprot_id
# Find the next available model number
model_id = max([m.id[0] for m in session.models], default = 0) + 1
# Open the overlapping component models.
models = []
fstep = (residues_per_file // overlap) - 1
for i in range(1,nfiles+1,fstep):
open_next_model(filename % i,
residues_per_file, overlap, align_span, models)
# Add last model which may overlap by a different number of residues.
if (nfiles-1) % fstep != 0:
last_overlap = overlap + overlap*(fstep - ((nfiles-1) % fstep))
open_next_model(filename % nfiles,
residues_per_file, last_overlap, align_span, models)
# Combine the segment models into one
model_ids = '#' + ','.join(m.id_string for m in models)
from chimerax.core.commands import run
if combine:
# Combine into one model
shift_residue_numbers(models)
copy = run(session, ('combine %s name %s modelId #%d close true'
% (model_ids, uniprot_id, model_id)))
models = [copy]
# TODO: Currently the combine command cannot preserve chain ids.
else:
# Group models under a parent model
run(session, 'rename %s %s id #%d' % (model_ids, uniprot_id, model_id))
# Adjust lighting and center view.
run(session, 'light full')
run(session, 'view')
return models
def open_next_model(path, residues_per_file, overlap, align_span, models):
from chimerax.core.commands import run
model = run(session, 'open %s' % path)[0]
id = model.id_string
run(session, 'color bfactor #%s palette alphafold' % id)
run(session, 'hide #%s cartoon ; show #%s atoms ; style #%s sphere' % (id,id,id))
end_match = '%d-%d' % (residues_per_file-5, residues_per_file-1) # 1395-1399
if models:
last_model = models[-1]
run(session, 'align #%s:%d-%d@CA to #%s:%s@CA'
% (model.id_string, overlap-align_span+1, overlap,
last_model.id_string, end_match))
run(session, 'delete #%s:1-%d' % (model.id_string, overlap))
models.append(model)
def shift_residue_numbers(structures):
# First adjust residue numbers of each segment
rnext = 1
for i,m in enumerate(structures):
rnums = m.residues.numbers
rmax, rmin = rnums.max(), rnums.min()
m.residues.numbers += rnext - rmin
rnext += rmax-rmin+1
def register_command(session):
from chimerax.core.commands import CmdDesc, register, StringArg, OpenFolderNameArg, BoolArg
desc = CmdDesc(required=[('uniprot_id', StringArg)],
keyword=[('directory', OpenFolderNameArg),
('combine', BoolArg)],
synopsis='Open multifile AlphaFold model')
register('bigalpha', desc, open_multifile_alphafold_model, logger=session.logger)
register_command(session)
Tom Goddard, September 8, 2021