ChimeraX Recipes
Average lipophilicity of alpha helices
Here is Python code defining a command “helixmlp” that computes the average lipophilicity for each alpha helix. The purpose was to color the helices by lipophilicity as a guide to which might be transmembrane. The average is computed for every atom of the alpha helix weighted by its solvent accessible surface area of the helix alone. The lipophilicity values are looked up for standard amino acids in the same way as the ChimeraX mlp command. The average value for each helix is assigned to all residues of the helix as the attribute named “helix_lipophilicity” and can be used to color the helix using the ChimeraX Render by Attribute tool or the equivalant color byattribute command.
To define the helixmlp command open helixmlp.py in ChimeraX
open helixmlp.py
then open a structure and use the helixmlp command on it and color using, for example,
open 8f76
helixmlp #1
preset cartoon cylinders
color byattribute r:helix_lipophilicity #1 palette lipophilicity
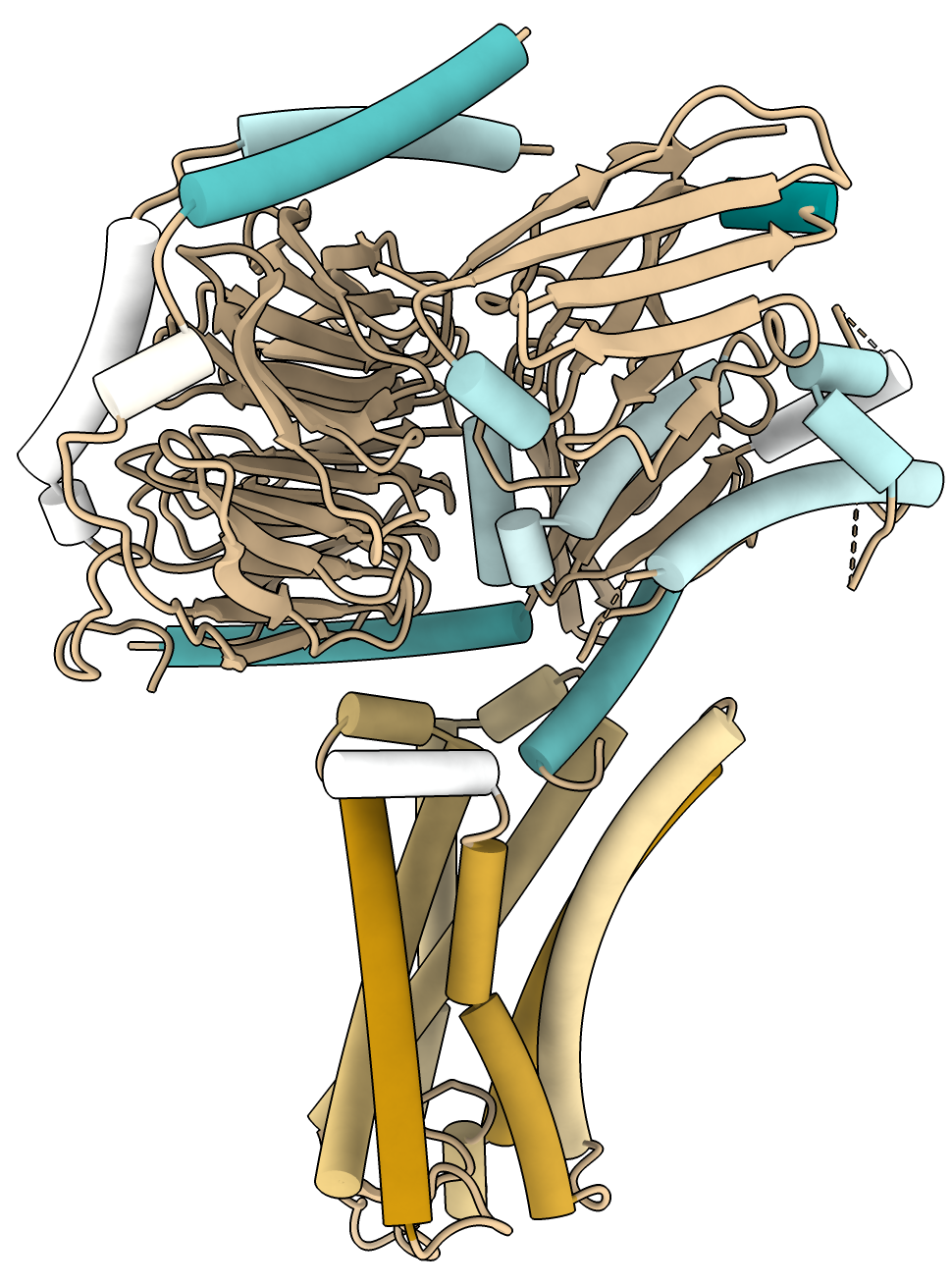
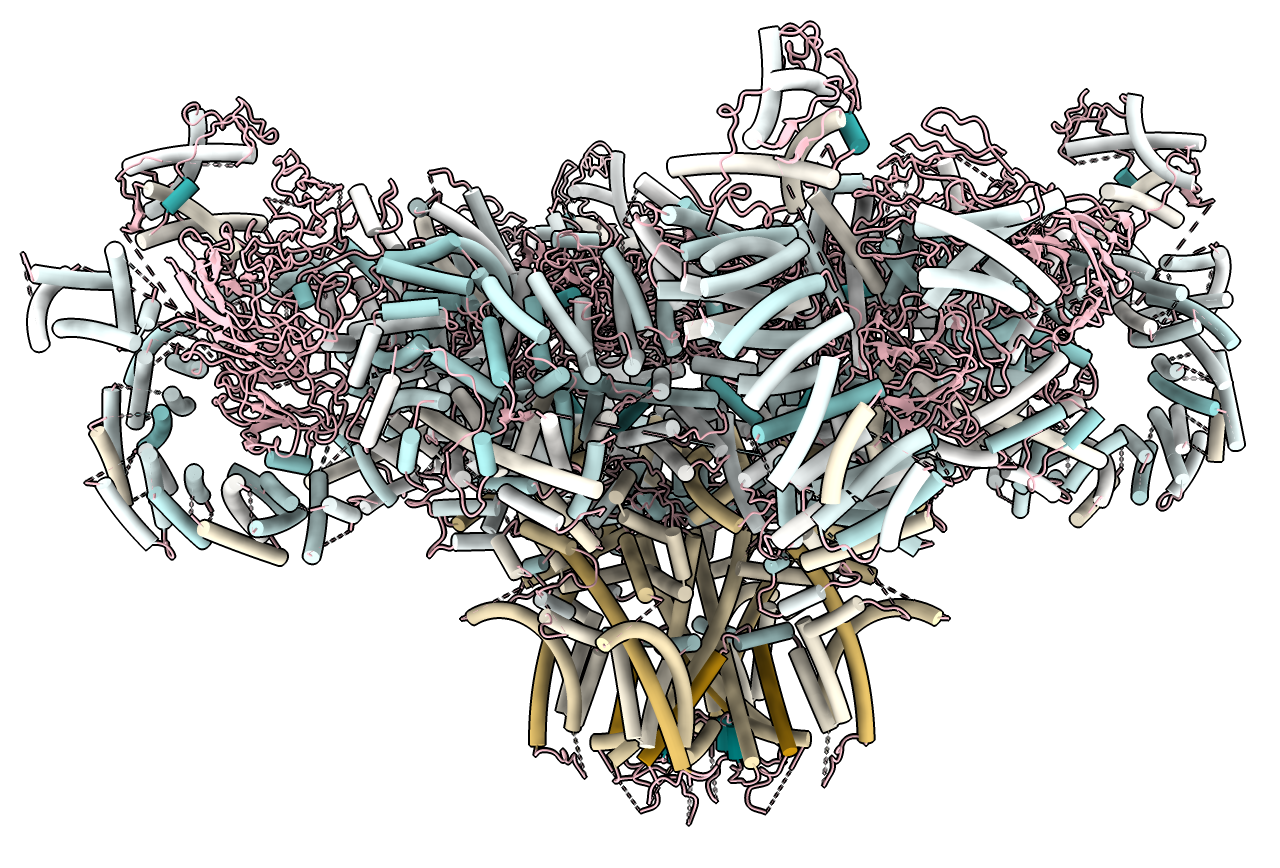
This shows a GPCR transmembrane protein. The orange helices are lipophilic and are in the lipid membrane while the blue helices are part of intracellular G proteins. Another example showing the rabbit ryanodine receptor PDB 5j8v similarly identifies the transmembrane helices.
Here is the helixmlp.py code:
# Color each helix to show how hydrophobic it is in order to highlight which helices
# might be transmembrane.
# To do this, for each atom in the helix consider its solvent accessible surface area
# for that lone helix and multiply by the mlp lipophilicity of that atom, sum over
# all atoms and divide by SAS area to get a area weighted lipophilicity value.
# Then color the helices by that value. Assign the average helix value to each
# atom and to each residue in the helix so render by attribute can do the coloring.
def sas_average_lipophilicity(session, residues):
atoms = residues.atoms
from chimerax.surface.measure_sasacmd import measure_sasa
measure_sasa(session, atoms)
lsum = 0
asum = 0
for a in atoms:
a_lip = atom_lipophilicity(a)
if a_lip is not None:
lsum += a.area * a_lip
asum += a.area
ave_lipo = lsum / asum if asum > 0 else 0
return ave_lipo
_lipophilicity_table = None
def atom_lipophilicity(atom):
global _lipophilicity_table
if _lipophilicity_table is None:
from chimerax.mlp.mlp import Defaults
lipophilicity = Defaults().fidatadefault
rname = atom.residue.name
if rname not in lipophilicity:
return None
l = lipophilicity[rname].get(atom.name)
return l
def helix_residues(residues):
hlist = []
hres = []
from chimerax.atomic import Residues
for r in residues:
if r.is_helix:
hres.append(r)
elif hres:
hlist.append(Residues(hres))
hres = []
if hres:
hlist.append(Residues(hres))
return hlist
def helix_lipophilicity(session, residues):
for hres in helix_residues(residues):
ave_lipo = sas_average_lipophilicity(session, hres)
for r in hres:
r.helix_lipophilicity = ave_lipo
for a in r.atoms:
a.helix_lipophilicity = ave_lipo
from chimerax.atomic import Residue, Atom
Residue.register_attr(session, 'helix_lipophilicity', 'Helix lipophilicity', attr_type = float)
Atom.register_attr(session, 'helix_lipophilicity', 'Helix lipophilicity', attr_type = float)
def register_command(logger):
from chimerax.core.commands import register, CmdDesc
from chimerax.atomic import ResiduesArg
desc = CmdDesc(required = [('residues', ResiduesArg)],
synopsis='Set helix_lipophilicity attribute')
register('helixmlp', desc, helix_lipophilicity, logger=logger)
register_command(session.logger)
Tom Goddard, March 7, 2024